Transforming¶
Operations on compounds are implemented as Transformers in
scikit-chem, which are analoguous to Transformer objects in
scikit-learn. These objects define a 1:1 mapping between input and
output objects in a collection (i.e. the length of the collection
remains the same during a transform). These mappings can be very varied,
but the three main types currently implemented in scikit-chem are
Standardizers, Forcefields and Featurizers.
Standardizers¶
Chemical data curation is a difficult concept, and data may be formatted differently depending on the source, or even the habits of the curator.
For example, solvents or salts might be included the representation, which might be considered an unnecessary detail to a modeller, or even irrelevant to an experimentalist, if the compound is solvated is a standard solvent during the protocol.
Even the structure of molecules that would be considered the ‘same’, can often be drawn very differently. For example, tautomers are arguably the same molecule in different conditions, and mesomers might be considered different aspects of the same molecule.
Often, it is sensible to canonicalize these compounds in a process called Standardization.
In scikit-chem, the
standardizers package provides
this functionality. Standardizer objects transform Mol objects
into other Mol objects, which have their representation
canonicalized (or into None if the protocol fails). The details of
the canonicalization may be configured at object initialization, or by
altering properties.
Tip
Currently, the only available Standardizer is a wrapper of the ChemAxon Standardizer. This requires the ChemAxon JChem software suite to be installed and licensed (free academic licenses are available from the website). We hope to implement an open source Standardizer in future.
As an example, we will standardize the sodium acetate:
In [3]:
mol = skchem.Mol.from_smiles('CC(=O)[O-].[Na+]', name='sodium acetate'); mol.to_smiles()
Out[3]:
'CC(=O)[O-].[Na+]'
A Standardizer object is initialized:
In [43]:
std = skchem.standardizers.ChemAxonStandardizer()
Calling transform on sodium acetate yields the conjugate ‘canonical’ acid, acetic acid.
In [44]:
mol_std = std.transform(mol); mol_std.to_smiles()
Out[44]:
'CC(=O)O'
The standardization of a collection of Mols can be achieved by
calling transform on a pandas.Series:
In [45]:
mols = skchem.read_smiles('https://archive.org/download/scikit-chem_example_files/example.smi',
name_column=1); mols
Out[45]:
name
ethane <Mol: CC>
propane <Mol: CCC>
benzene <Mol: c1ccccc1>
sodium acetate <Mol: CC(=O)[O-].[Na+]>
serine <Mol: NC(CO)C(=O)O>
Name: structure, dtype: object
In [46]:
std.transform(mols)
ChemAxonStandardizer: 100% (5 of 5) |##########################################| Elapsed Time: 0:00:01 Time: 0:00:01
Out[46]:
name
ethane <Mol: CC>
propane <Mol: CCC>
benzene <Mol: c1ccccc1>
sodium acetate <Mol: CC(=O)O>
serine <Mol: NC(CO)C(=O)O>
Name: structure, dtype: object
A loading bar is provided by default, although this can be disabled by lowering the verbosity:
In [47]:
std.verbose = 0
std.transform(mols)
Out[47]:
name
ethane <Mol: CC>
propane <Mol: CCC>
benzene <Mol: c1ccccc1>
sodium acetate <Mol: CC(=O)O>
serine <Mol: NC(CO)C(=O)O>
Name: structure, dtype: object
Forcefields¶
Often the three dimensional structure of a compound is of relevance, but many chemical formats, such as SMILES do not encode this information (and often even in formats which serialize geometry only coordinates in two dimensions are provided).
To produce a reasonable three dimensional conformer, a compound must be roughly embedded in three dimensions according to local geometrical constraints, and forcefields used to optimize the geometry of a compound.
In scikit-chem, the forcefields
package provides access to this functionality. Two forcefields, the
Universal Force Field
(UFF) and the Merck
Molecular Force Field (MMFF) are currently provided. We will use the
UFF:
In [23]:
uff = skchem.forcefields.UFF()
mol = uff.transform(mol_std)
In [25]:
mol.atoms
Out[25]:
<AtomView values="['C', 'C', 'O', 'O', 'H', 'H', 'H', 'H']" at 0x12102b6a0>
This uses the forcefield to generate a reasonable three dimensional
structure. In rdkit (and thus scikit-chem, conformers are
separate entities). The forcefield creates a new conformer on the
object:
In [27]:
mol.conformers[0].atom_positions
Out[27]:
[<Point3D coords="(1.22, -0.48, 0.10)" at 0x1214de3d8>,
<Point3D coords="(0.00, 0.10, -0.54)" at 0x1214de098>,
<Point3D coords="(0.06, 1.22, -1.11)" at 0x1214de168>,
<Point3D coords="(-1.20, -0.60, -0.53)" at 0x1214de100>,
<Point3D coords="(1.02, -0.64, 1.18)" at 0x1214de238>,
<Point3D coords="(1.47, -1.45, -0.37)" at 0x1214de1d0>,
<Point3D coords="(2.08, 0.21, -0.00)" at 0x1214de2a0>,
<Point3D coords="(-1.27, -1.51, -0.08)" at 0x1214de308>]
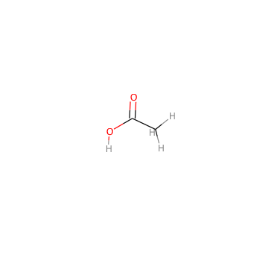
The molecule can be visualized by drawing it:
In [35]:
skchem.vis.draw(mol)
Out[35]:
<matplotlib.image.AxesImage at 0x1236c6978>
Featurizers (fingerprint and descriptor generators)¶
Chemical representation is not by itself very amenable to data analysis and mining techniques. Often, a fixed length vector representation is required. This is achieved by calculating features from the chemical representation.
In scikit-chem, this is provided by the descriptors package. A
selection of features are available:
In [11]:
skchem.descriptors.__all__
Out[11]:
['PhysicochemicalFeaturizer',
'AtomFeaturizer',
'AtomPairFeaturizer',
'MorganFeaturizer',
'MACCSFeaturizer',
'TopologicalTorsionFeaturizer',
'RDKFeaturizer',
'ErGFeaturizer',
'ConnectivityInvariantsFeaturizer',
'FeatureInvariantsFeaturizer',
'ChemAxonNMRPredictor',
'ChemAxonFeaturizer',
'ChemAxonAtomFeaturizer',
'GraphDistanceTransformer',
'SpacialDistanceTransformer']
Circular fingerprints (of which Morgan fingerprints are an example) are often considered the most consistently well performing descriptor across a wide variety of compounds.
In [12]:
mf = skchem.descriptors.MorganFeaturizer()
mf.transform(mol)
Out[12]:
morgan_fp_idx
0 0
1 0
2 0
3 0
4 0
..
2043 0
2044 0
2045 0
2046 0
2047 0
Name: MorganFeaturizer, dtype: uint8
We can also call the standardizer on a series of Mols:
In [13]:
mf.transform(mols.structure)
MorganFeaturizer: 100% (5 of 5) |##############################################| Elapsed Time: 0:00:00 Time: 0:00:00
Out[13]:
| morgan_fp_idx | 0 | 1 | 2 | 3 | 4 | ... | 2043 | 2044 | 2045 | 2046 | 2047 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 0 | 0 | 0 | 0 | 0 | ... | 0 | 0 | 0 | 0 | 0 |
| 1 | 0 | 0 | 0 | 0 | 0 | ... | 0 | 0 | 0 | 0 | 0 |
| 2 | 0 | 0 | 0 | 0 | 0 | ... | 0 | 0 | 0 | 0 | 0 |
| 3 | 0 | 0 | 0 | 0 | 0 | ... | 0 | 0 | 0 | 0 | 0 |
| 4 | 0 | 1 | 0 | 0 | 0 | ... | 0 | 0 | 0 | 0 | 0 |
5 rows × 2048 columns
Note
Note that Morgan fingerprints are 1D, and thus when we use a single Mol as input, we get the features in a 1D pandas.Series . When we use a collection of Mol s, the features are returned in a pandas.DataFrame , which is one higher dimension than a pandas.Series, as a collection of Mol s are a dimension higher than a Mol by itself.
Some descriptors, such as the AtomFeaturizer , will yield 2D features when used on a Mol, and thus will yield the 3D pandas.Panel when used on a collection of Mol s.